バイオインフォマティクスで大枠を学んだが、遺伝子の差異によって、人がサルから進化したし、人の中でも人種の違い、それぞれの個人の特徴なども遺伝子の違いから生じている。先天的なものだけでなく、がんの発症など疾病に関しても遺伝子の変化が影響している。遺伝子の違いをどのように特定していくかというところをまとめてみた。
ヒトゲノムの構造
人にはタンパク質を生成する遺伝子が22,000個ある。ゲノムを構成する塩基対が30億個あることを考えると、タンパク質生成に関連している遺伝子はごくわずかしかない。実際には転写されたときに選択的スプライシング(mRNAに転写する際にイントロンという使わない部分を除去して、エキソンだけの成熟mRNAにする過程)にもパターンがあり、転写物にバリエーションを生み出している。またタンパク質を生成しない部分のゲノムにも何かしら機能があると考えられている。
バリアントとは
それでは、生物種ごと、人種ごと、個人ごとの違いを理解するには、遺伝子やゲノムがどのように違ってくるのかを調べる必要がある。バリアントとは遺伝子の多様性を意味する言葉であり、病気に関係する病的変異と、病気に関係しない遺伝子多型が含まれる。
多型の種類としては、一塩基の違いによるもの(SNP)や、挿入や欠損によるもの(Indel)、塩基配列の逆転、配列のコピー数が異なるもの(CNV)など、いくつかの種類がある。SNPやIndelはショートバリアントと呼ばれる。
バリアントコール
バリアントを検出する際にReferenceとなる参考配列と異なる部分を検出することをバリアントコールと呼ぶ。バリアントコールを学ぶために、UCLAのYoutubeの動画があったので、こちらを元に整理してみる。遺伝子多型は、生殖細胞と体細胞の両方で発生するが、この動画では生殖細胞に絞って、GATKを使ったレクチャーを行なっている。GATKはショートバリアントを検出して、VCFフォーマットで出力するプログラムであり、以下のフローでバリアントを検出する。
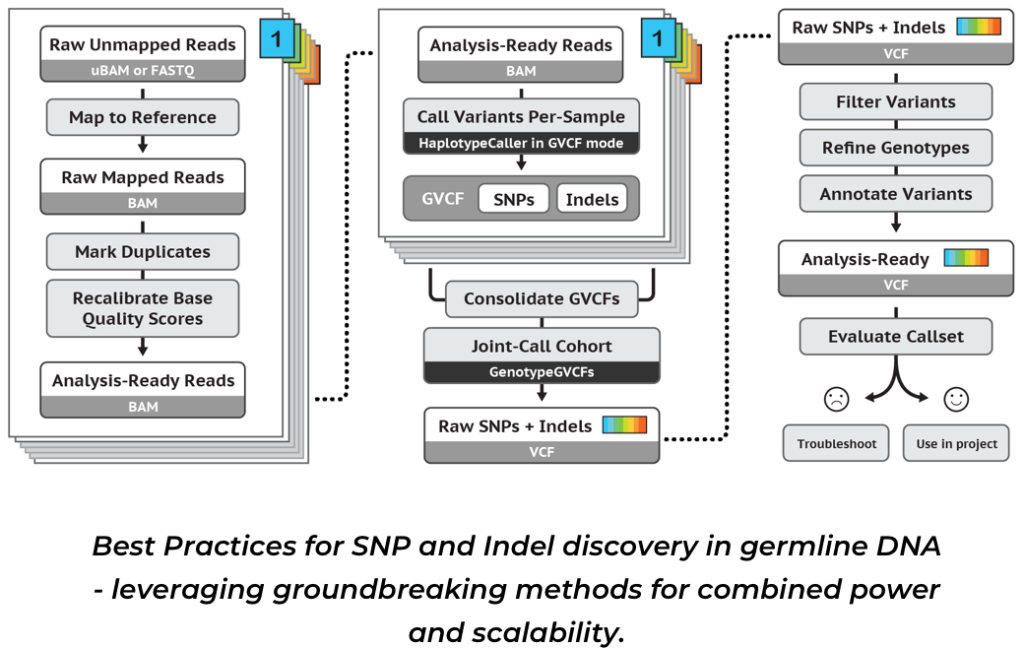
バリアントコールの4つのステップ
次世代シーケンサー(NGS)で得られたリードを以下の手順で処理していく。
- NGSで読み取ったリードのFASTQファイルを参照ゲノムにマッピングし、BAMファイルを作成する。FASTQはFASTAデータに対して、リード情報に加えて塩基ごとのクオリティスコアが付加されている。
- BAMファイルをクリーニングする。これはいわゆるデータの前処理にあたる。複製されている部分の削除やBQSR(Base Quality Score Recalibration)による品質スコアの調整行う。BQSRは既知のバリアントデータをもとに機械学習を使って、入力データの品質スコアに含まれるエラーを調整するプロセス。
- GATKを使い、BAMファイルからGVCF(Genomic Variant Call Format)を作成する。VCFがバリアントのある場所のみが保存されるのに対し、このGVCFはゲノム上の全てのポジションのレコードを含んでいる。このステップで、ハプロタイプを推定し、それをもとにバリアントを検出する。
- コホート(特定の集団)内の全ての情報を統合し、VCFを作成する。
生殖細胞と体細胞との違い
生殖細胞のバリアントコールについて説明したが、体細胞はいくつかの理由から難易度が高い。まず最初に生殖細胞はリファレンス配列との比較だったが、体細胞で例えばがんにおける変異を特定するには、正常細胞との違いも考える必要がある。また、個人とコホート内との差異なども必要で、生殖細胞とは違った点が多く、上記の手法は使えないとのこと。これについては別途調べていきたい。